 J Clin Aesthet Dermatol. 2023;16(1):25-29.
J Clin Aesthet Dermatol. 2023;16(1):25-29.
by Landon K. Hobbs, MD; Merrick D. Kozak, MD; and Alejandro A. Gru, MD
All authors are with the Department of Pathology at the University of Virginia School of Medicine in Charlottesville, Virginia.
ABSTRACT: Background. Sclerodermiform lupus erythematosus (SDLE) is a rare Type 3 overlap syndrome of morphea and cutaneous lupus diagnosed with histopathologic features of both diseases present. It was first reported in 1976 by Umbert et al with a case series of four patients. SDLE is more common in young to middle-aged female patients.
Methods. After IRB approval, we searched our internal pathology database for cases of SDLE, excluded any patients that did not fulfill the diagnostic criteria, and verified each case with a board-certified dermatopathologist.
Results. Five patients with SDLE were identified; three of the patients are male and three are Black. Consistent with prior reports in the literature, the lesions were described as hyperpigmented plaques or nodules. The most commonly involved location was the extremities. Histopathology showed diffuse sclerosis in all five patients’ biopsies, vacuolar interface changes in three biopsies, basement membrane thickening in one biopsy, and increased dermal mucin deposition in two of the four biopsy specimens stained with colloidal iron. Improvement was noted in patients treated with topical, intralesional, or systemic corticosteroids, topical calcineurin inhibitors, and oral antimalarials.
Conclusion. We describe five cases of SDLE which is the largest series to date and the first series with a majority of patients being male. Improved recognition and a more thorough understanding of SDLE is necessary for appropriate diagnosis and management.
Keywords: Sclerodermiform lupus erythematosus, cutaneous lupus erythematosus, morphea, overlap syndromes.
Sclerodermiform lupus erythematosus (SDLE), or sclerodermiform linear lupus erythematosus when the lesions appear linear, is a rare Type 3 overlap syndrome between morphea and chronic cutaneous lupus erythematosus (CCLE) such as discoid lupus erythematosus (DLE) or lupus panniculitis/profundus (LEP). Overlap syndromes are disorders that satisfy diagnostic criteria of two or more different diseases concurrently or consecutively, with Type 3 being between two cutaneous-only diseases, Type 1 indicating overlapping systemic diseases, and Type 2 meaning a systemic disease overlapping with a cutaneous disease.1,2 It has also been hypothesized that SDLE may represent a distinct clinical condition.3 The diagnostic criteria is overlapping histopathologic features of both CCLE and morphea. SDLE is more common in young adults and adolescent females, however cases in males have been reported.1,2,4 Since Umbert et al first suggested the term “sclerodermiform linear lupus erythematosus” in 1976, several cases have been reported, but the overall incidence appears to be low with the largest series to date being the four patients in that initial report.1–4 Here we report a series of five patients diagnosed with SDLE at a tertiary academic medical center since 2008. This is the largest case series of SDLE to date and the first series with a majority of male patients seen.
Methods
After obtaining IRB approval, we searched our internal pathology database of cases that mentioned “morpheaform” and “lupus”, “sclerodermiform” and “lupus”, “sclerodermoid” and “lupus” in the diagnostic line between 1/1/2000 and 6/1/2021. We excluded any patients that did not fulfill the diagnostic criteria of SDLE. For those patients treated at our institution, a comprehensive chart review was performed in order to obtain demographics, clinical features of initial presentation, histopathological findings, serological findings, and treatments with outcomes. For patients treated by an outside dermatologist, maximal efforts were made to obtain the above information. All cases were reviewed by a board-certified dermatopathologist to ensure the appropriate diagnosis.
Using PubMed, we searched “morpheaform lupus”, “sclerodermiform linear lupus erythematosus,” “sclerodermiform lupus,” and “sclerodermoid lupus” to find case reports and series. Results were filtered for human species and English language. Additional reports were found by scanning references of the located papers as well the papers that had cited them. Using the cases found, we evaluated demographic, clinical, serological, and histopathological information to compare to our case series.
Case Series
Three patients with SDLE were diagnosed recently on the referral consultation practice, and three additional cases mentioning “morpheaform/sclerodermiform/sclerodermoid” and “lupus” in the diagnostic line or microscopic description were located via an internal pathology database search from 1/1/2000 and 6/1/2021. However, after a review by the board-certified dermatopathologist, one was excluded because the lesions of morphea and lupus were in separate anatomical locations. The other five cases were verified to be SDLE and are included in this case-series (Table 1).
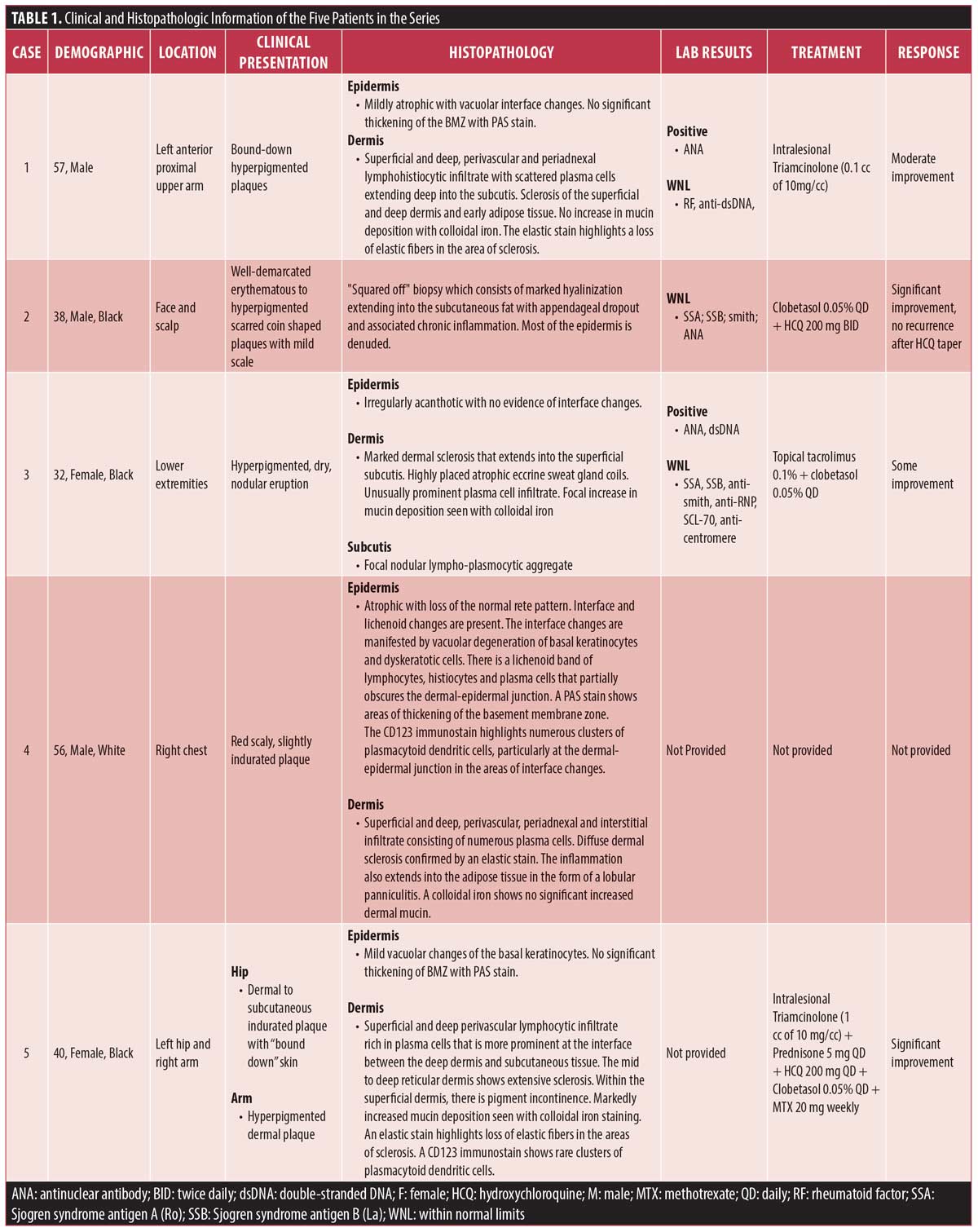
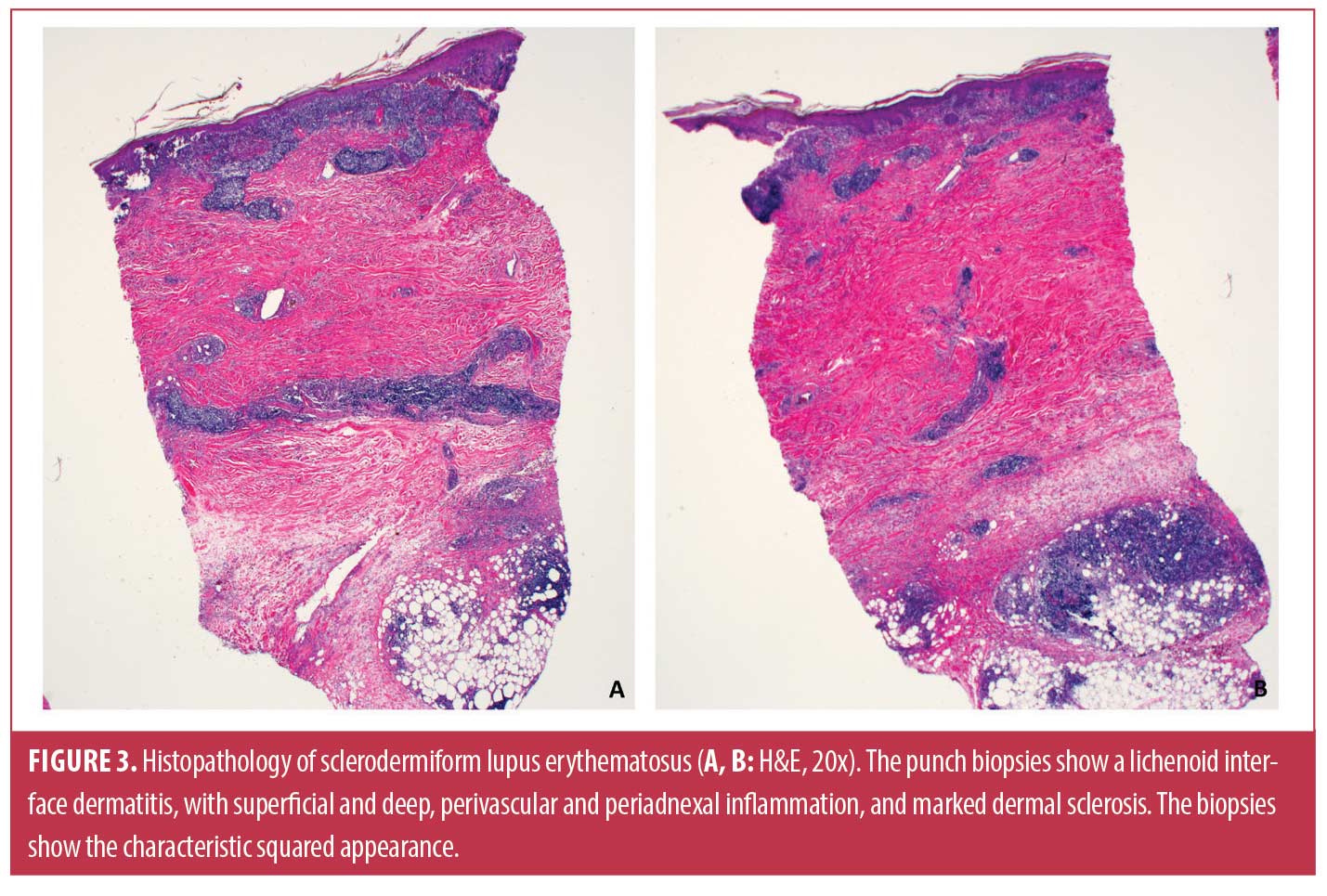
In our series of five patients, three (60%) were male, three (60%) were Black, and the mean age at presentation was 44.6 years. The most commonly involved location was the extremities. Clinically, the SDLE lesions were described as hyperpigmented plaques or nodules (Figure 1). Two patients were also noted to have bound-down skin (Figure 2). Histopathology (Figures 3–6) showed diffuse sclerosis in all five patients, with three (60%) having involvement of the subcutaneous tissue. An atrophic or denuded epidermis was seen in three (60%) patients. Vacuolar interface changes were present in three of the biopsies (60%). Periodic acid-Schiff (PAS) staining was performed on specimens from three patients and only showed basement membrane thickening in one (33%). Increased dermal mucin deposition was seen in two of the four (50%) biopsy specimens stained with colloidal iron. Direct immunofluorescence (DIF) was not performed in any of the cases. ANA was positive in 66.7 percent (2/3), while anti-dsDNA was positive in 50 percent (1/2) of tested patients, respectively. Clobetasol was the most common treatment (3 patients); all three patients had improvement in their lesions. Improvement was also noted in patients treated with intralesional triamcinolone (2 patients), oral hydroxychloroquine (2 patients), topical tacrolimus (1 patient), and oral prednisone (1 patient).





Discussion
Clinically, sclerodermiform LE may present as either morphea or CCLE, however most cases have a morphea-like appearance. Typically, the patients present with red-brown plaques with an atrophic center.1,3 The arms are the most commonly involved location, followed by the face and scalp, trunk, then buttocks and thighs. The results of this series were consistent with prior case reports, including involvement of the lower extremities. Diagnosis can be difficult, as the skin lesions of the two diseases have been reported to occur at separate locations or as coexistent disease in the same location.2 However, we do not believe separate anatomical locations constitute a true diagnosis of SDLE. At our institution, we found a similar case and excluded it in this report as mentioned in the results. Although some patients have been found to have a positive ANA, anti-dsDNA, or SS-A, no specific trend has been reported in regards to laboratory values.2,5 Such low prevalence of antibodies is compatible with what is typically observed in cases of CCLE.
The histopathology of SDLE shows characteristics of both morphea and CCLE, either concurrently or consecutively in the same anatomical lesion. Typical findings include interface dermatitis with hydropic degeneration of the basal cell layer, basement membrane zone thickening, and increased dermal mucin, characteristic of CCLE, in addition to excessive collagen deposition leading to thickening of the dermis and/or subcutaneous tissues, which is characteristic of morphea.1,3,6–8 Perivascular inflammation is also a common finding.6,8,9 Our series was consistent with these reported histopathologic findings as all five patients’ biopsies demonstrating increased collagen deposition, three (60%) demonstrating an interface dermatitis, and two having increased dermal mucin. Isolated case studies have shown positive DIF findings with deposition of IgG, IgM, and/or C3 along the dermal-epidermal junction.2,3
The differential diagnosis includes different sclerosing disorders such as mixed connective tissue disease (MCTD), morphea/scleroderma, LEP, and overlap of morphea with lichen sclerosus. MCTD was considered, however none of the patients exhibited significant systemic symptoms consistent with MCTD and only one patient had anti-RNP testing performed. Histopathologically, morphea typically shows extensive sclerosis involving the deeper dermis and interlobular fat septa but an interface reaction pattern is not identified.1,6,7,9 However, some cases of morphea can show histopathologic findings of lichen sclerosus in the surface, which might include interface changes. The presence of superficial dermal edema (typical of lichen sclerosus), lack of deep perivascular and periadnexal inflammation, and clinical appearance of the lesions, might help to differentiate from SDLE. As opposed to SDLE, deep periadnexal inflammation is less common in morphea and LS. The typical histopathologic findings of LEP include lobular lymphocytic panniculitis with germinal centers often with associated hyaline sclerosis and/or fat necrosis.6,7,10 However, unlike in cases of SDLE, diffuse dermal sclerosis is not seen. Our five cases all had extensive dermal sclerosis and no germinal centers within the subcutis which distinguishes them from LEP. Scleroderma and LEP can have many overlapping features such as lymphocytic panniculitis, lymphoid nodular structures in the fat, broadening of fibrous septa, and lymphocytic vasculitis, however hyaline necrosis of fat favors a diagnosis of LEP even in specimens lacking characteristic changes of lupus.11 CD34, smooth muscle actin (SMA), and myxovirus resistance protein 1 (MXA) have been reported to help distinguish LEP, scleroderma, and morphea.7 Lichen sclerosus is characterized by follicular plugging, hyperkeratosis, epidermal atrophy, basal cell hydropic degeneration, and homogenized papillary dermis.12 Some cases can also show the follicular plugging characteristic of DLE.
Commonly reported treatment regimens include antimalarials, topical corticosteroids or calcineurin inhibitors, or systemic corticosteroids.1,2 Systemic corticosteroids were only used in one patient in our series. Treatment and outcome data was not provided for one patient, however the four patients with treatment data were noted to have improvement of their lesions. Antimalarials, topical corticosteroids, topical calcineurin inhibitors, as well as intralesional triamcinolone were seen to be effective. It has recently been reported that ultrasonography may be a useful noninvasive tool for monitoring therapeutic response and for modifying treatment in patients with SDLE.5
Conclusion
We describe five new cases of SDLE, the largest series to date, and integrate our findings with those in the literature. Although reported to have a significant female predominance, our series shows that SDLE is diagnosed in males as well. Improved recognition and a more thorough understanding of SDLE is necessary for appropriate management, as well as a better communication with the patient about complications and prognosis of their disease.
References
- Khelifa E, Masouye I, Pham HC, et al. Linear sclerodermic lupus erythematosus, a distinct variant of linear morphea and chronic cutaneous lupus erythematous. Int J Dermatol. 2011;50(12):1491–1495.
- Pascucci A, Lynch PJ, Fazel N. Lupus erythematosus and localized scleroderma coexistent at the same sites: a rare presentation of overlap syndrome of connective-tissue diseases. Cutis. 2016;97(5):359–363.
- Julià M, Mascaró JM, Guilabert A, et al. Sclerodermiform linear lupus erythematosus: A distinct entity or coexistence of two autoimmune diseases? J Am Acad Dermatol. 2008;58(4):665–667.
- Umbert P, Winkelmann RK. Concurrent localized scleroderma and discoid lupus erythematosus. Cutaneous “mixed” or “overlap” syndrome. Arch Dermatol. 1978;114(10):1473–1478.
- Cheng CY, Huang YL, Lee MC, et al. Ultrasonography for assessing the disease activity of sclerodermoid lupus erythematosus panniculitis. Indian J Dermatol Venereol Leprol. 2021;87(1):146–146.
- Elbendary A, Griffin J, Li S, et al. Linear Sclerodermoid Lupus Erythematosus Profundus in a Child. Am J Dermatopathol. 2016;38(12):904–909.
- Schwartz Z, Magro CM. Intralesional overlap syndrome: Sclerodermic lupus panniculitis and sclerodermic discoid lupus erythematosus. JAAD Case Rep. 2020;6(3):166–168.
- Yu Y, Liu CY, Chan J, et al. Sclerodermiform lupus erythematosus: A rare case presented as cicatricial alopecia. Dermatol Sin. Published online 2015.
- Bernárdez C, Prieto-Torres L, Macías E, et al. Concurrent presentation of cutaneous lesions of deep linear morphoea and discoid lupus erythematosus. Lupus. 2016;25(2):204–208.
- Marzano AV, Tanzi C, Caputo R, et al. Sclerodermic linear lupus panniculitis: report of two cases. Dermatol Basel Switz. 2005;210(4):329–332.
- Stork J, Vosmík F. Lupus erythematosus panniculitis with morphea-like lesions. Clin Exp Dermatol. 1994;19(1):79–82.
- Almuqati RR, Hariri J, Abduljabbar M. Histopathological Coexistence of Extragenital Lichen Sclerosus and Morphea in a Single Lesion. Cureus. 12(12):e12215.