J Clin Aesthet Dermatol. 2019;12(5):28–32
 by Raymond E. Kleinfelder, DO; Nady Hin, DO; Stefanie Cubelli, DO; Allan Snyder, PA-C; Francisco Kerdel, BSc, MBBS; and Brad Glick, DO, MPH
by Raymond E. Kleinfelder, DO; Nady Hin, DO; Stefanie Cubelli, DO; Allan Snyder, PA-C; Francisco Kerdel, BSc, MBBS; and Brad Glick, DO, MPH
Drs. Kleinfelder and Hin are with the Dermatology Residency Program at the LECOMT/Larkin Community Hospital, Palm Springs Campus, in Hialeah, Florida. Dr. Cubelli is with St. John’s Episcopal Hospital in Far Rockaway, New York. Mr. Snyder is with Glick Skin Institute in Margate, Florida. Dr. Kerdel is with the the Dermatology Residency Program at the LECOMT/Larkin Community Hospital, Palm Springs Campus in Hialeah, Florida; the Florida Academic Dermatology Center in Coral Gables, Florida; and Florida International University in Miami, Florida. Dr. Glick is with the Dermatology Residency Program at the LECOMT/Larkin Community Hospital, Palm Springs Campus, in Hialeah, Florida and the Glick Skin Institute in Margate, Florida.
FUNDING: No funding was received for this study.
DISCLOSURES: Dr. Kerdel and Dr. Glick are speakers/have received honoraria from AbbVie, the manufacturer of adalimumab. The other authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Pyoderma gangrenosum (PG) is a rare, ulcerative, inflammatory skin disease that most commonly presents on the lower legs. Development of PG on the head and neck is exceedingly rare. We present the case of a 52-year-old man with no known history of underlying systemic disease who developed multiple facial lesions of PG that were refractory to both standard and alternative treatment modalities. Clearance of disease was ultimately achieved using adalimumab.
KEYWORDS: Adalimumab, atypical pyoderma gangrenosum, facial pyoderma gangrenosum, pyoderma gangrenosum, TNF-alpha inhibitor
Pyoderma gangrenosum (PG) is a rare, ulcerative, inflammatory skin disease that most commonly presents on the lower legs of middle-aged Caucasian women.1,2 PG is historically classified as a neutrophilic dermatosis.1 New insights into the pathophysiology of the disease demonstrate autoreactive T-cells and aberrant production of cytokines, such as tumor necrosis factor alpha (TNF-alpha), interleukin (IL)-1-beta, IL-17, and IL-23.1,3,4 This complex, autoinflammatory etiology might precede neutrophilic migration.1,4 While PG might be idiopathic, at least half of patients have underlying systemic comorbidities, with inflammatory bowel disease (IBD), rheumatoid arthritis, and hematologic disorders and malignancies being the most common.2,3,5 Additionally, PG-associated genetic syndromes further aid in our understanding of the autoinflammatory nature of the disease.1,4
Four main variants of PG have been described: ulcerative, pustular, bullous, and vegetative.6 The ulcerative type is the most common, and classically presents as a sterile pustule that progressively expands into an intensely painful ulcer with violaceous, undermined borders, and heals with cribriform scarring.2,7 While the ulcerative and pustular variants are typically found in patients with IBD, the bullous type most frequently occurs in patients with hematological malignancies and carries the worst prognosis.5,6 The vegetative or superficial subtype is more common in patients with no underlying comorbidities, and portends the best prognosis.8
Pathergy is an important triggering factor that occurs in approximately 30 percent of PG lesions; therefore, care must be taken when performing biopsies.2,7 While the histopathology for PG is not specific, it is important to rule out other infectious, neoplastic, or vasculopathic causes of ulceration.8 The differential diagnosis expands for presentations in atypical locations. The development of PG on the head and neck is exceedingly rare, and it is essential to rule out underlying granulomatosis with polyangiitis (previously called Wegener’s granulomatosis) in these patients.2,6,7 Ultimately, PG is a diagnosis of exclusion.2,7
The management of PG is complicated and must include a multimodal approach involving local wound care and the avoidance of trauma.9 There are currently no United States Food and Drug Administration (FDA)-approved medications for the treatment of PG.10 Systemic therapy is often necessary, with systemic corticosteroids and cyclosporine being the mainstays of therapy.11 PG is notorious for being refractory to treatment, and many patients require an alternative immunomodulator.9,11 With new insights into the pathogenesis of PG, biologic medications, such as TNF-alpha inhibitors, have become invaluable tools in cases of recalcitrant PG.9
Here, we present a case of multiple facial lesions of PG that was refractory to both standard and alternative treatment modalities in a patient with no known history of underlying systemic disease. Clearance of the disease was ultimately achieved using adalimumab (Humira®, AbbVie, Inc., Chicago, Illinois).
Case Presentation
A 52-year-old man presented to the outpatient clinic with a one-month history of an ulcer on the left inferior cutaneous lip (Figure 1). The patient had failed treatment with topical corticosteroids, antibiotics, and antifungals prescribed by a previous dermatologist. A punch biopsy performed one week prior to presentation demonstrated an ulcer with a dense collection of dermal neutrophils, minimal leukocytoclasia, and no definitive vasculitis. Biopsy was consistent with PG.
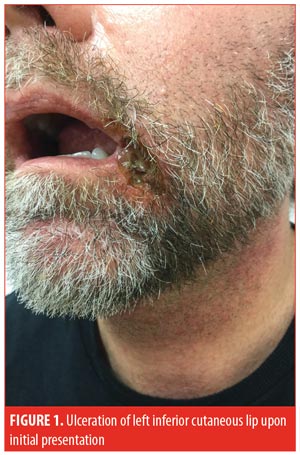
The patient had a history of gout and diverticular disease, which required partial resection of the sigmoid colon. The patient also reported a recent intentional 30-pound weight loss, achieved through diet and exercise. The patient’s weight upon presentation was 90 kilograms. The patient had no known history of IBD or malignancy, and colonoscopy results from three months prior showed no evidence of IBD. Complete blood count showed a mildly elevated white blood cell count, and CMP was normal. All antineutrophilic cytoplasmic antibodies (ANCAs) were negative. Serum protein electrophoresis and quantitative immunoglobulin levels failed to identify any significant abnormalities. Rheumatoid factor, anticyclic citrullinated peptide, and antinuclear antibody were negative. Initial bacterial and fungal swab cultures from the base of the ulcer showed no growth of organisms. Hepatitis B and C, human immunodeficiency virus (HIV), and QuantiFERON-TB Gold (Qiagen, Hilden, Germany) studies were negative.
After ruling out underlying disorders, the patient was prescribed clobetasol 0.05% paste, to apply to the base of the ulcer, and tacrolimus ointment, for the surrounding skin, as well as a 1mL intramuscular dose of triamcinolone (40mg/mL). After six weeks, the patient began to develop a new submental erosion, prompting the initiation of cyclosporine 100mg twice daily. After four additional weeks of no improvement, the cyclosporine was increased to 100mg three times daily and an oral corticosteroid (prednisone 20mg daily) was added to the regimen. The patient’s level of pain also required treatment with oral opioids.
Three months after the initial presentation, the patient developed a third ulceration on the right cheek, with concomitant worsening of the submental and lower lip ulcerations. Aerobic tissue cultures obtained via punch biopsy of the submental ulcer were positive for heavy growth of Escherichia coli and Enterococcus faecalis and moderate growth of Klebsiella oxytoca. Anaerobic tissue culture from punch biopsy demonstrated moderate growth of Propionibacterium acnes. Recommendation was made for inpatient hospitalization. The patient was hospitalized for five days, during which time, he received 1g of methylprednisolone daily, intravenous cefazolin, 750mg of ciprofloxacin twice daily, silver sulfadiazine ointment twice daily, and was continued on 100mg cyclosporine three times daily. During his hospitalization, the patient reported headache and chest pain and refused further cyclosporine. Upon discharge, the patient was started on methotrexate 15mg weekly, prednisone 20mg daily, and topical therapy.
The patient initially improved following hospital discharge; however, approximately one month post-discharge, the patient returned to the clinic with worsening of all three lesions. Thalidomide 100mg daily was initiated, prednisone was increased to 30mg daily, and methotrexate was continued. At the patient’s next one-month follow-up visit, he complained of even further worsening of all three ulcerations (Figure 2) and progressively increasing lower-extremity edema, prompting discontinuation of the thalidomide.
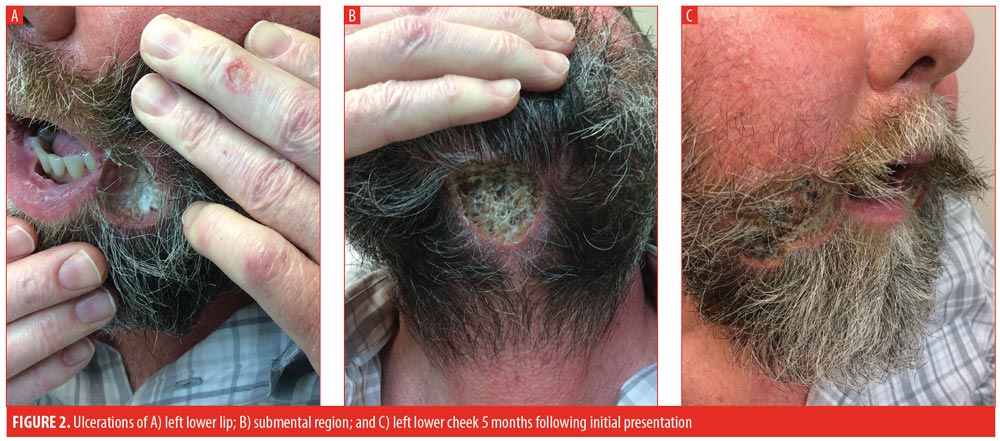
Three months postdischarge, the patient’s condition had not improved. After having failed numerous systemic therapies, adalimumab was selected for initiation. Prior to administration, the patient was sent for a QuantiFERON-TB Gold test to rule out tuberculosis; the test was negative. The patient was given an initial loading dose of 160mg, injected subcutaneously, followed by 80mg on Day 15, 40mg on Day 29, and 40mg weekly thereafter. The patient’s topical regimen was trolamine salicylate topical emulsion twice daily, cadexomer iodine gel twice daily, and tacrolimus ointment twice daily. The patient was seen at monthly intervals, during which time he began to stabilize. After two months of weekly adalimumab, sulfamethoxazole/trimethoprim 800mg/160mg daily was added to the regimen due to a concern about superinfection preventing healing. The patient was seen at regular intervals and exhibited progressive healing with weekly adalimumab. Monitoring included an annual QuantiFERON-TB Gold test. After 15 months of weekly adalimumab, all three lesions were almost completely healed and dosing was spaced to every other week. The patient’s most recent visit was during his 18th month of taking adalimumab, during which all three lesions of PG had completely resolved (Figure 3). To date, the patient has had no recurrence of the lesions. Adalimumab will be discontinued after one year of being disease-free and in the absence of indicated comorbidities.

Discussion
Facial PG is exceedingly rare. According to the largest retrospective analyses on PG, only 4.5 percent of 356 patients in one study and 7.8 percent of 103 patients in another study developed PG on the head or neck.2,7 Before accepting a diagnosis of PG on the head, neck, or oral cavity, it is essential that appropriate tests are performed to exclude granulomatosis with polyangiitis.6,12 ANCA studies in our patient were negative, and biopsy suggested no underlying vasculitis, confirming the diagnosis of PG. Kurniadi et al13 suggested that patients with an underlying comorbid disease are more likely to develop classic PG of the leg, whereas patients without an underlying disease, such as our patient, are more likely to develop lesions on the face. Kratzsch et al14 also described two patients with facial PG who were especially atypical due to their advanced age (89 and 90 years), nearly double the average age of a PG patient. Finally, in Japan, Takayasu arteritis is a common comorbidity in patients with PG, and 42.9 percent of these patients present with lesions on the head and neck.15
Classification of our patient into Powell’s described variants of PG6 is challenging. Our patient did not have IBD, nor did the location correspond to the classic locations of ulcerative or pustular PG.6 Bullous or atypical PG can present on the face, but is frequently linked to myeloproliferative disorders, which were ruled out in our patient. Vegetative or superficial granulomatous PG presents as erosions and superficial ulcers in patients with no underlying disease, yet typically responds well to treatment, unlike our recalcitrant patient.6,8
It is commonly stated that half of cases of PG are associated with an underlying systemic disease.3 More recent comprehensive reviews suggest that 50 to 70 percent might be a more appropriate number.2,7 It is essential to rule out comorbid disease, as treatment of these entities can be instrumental in the resolution of PG. IBD is the most common underlying disease in PG, and PG remains the most common cutaneous manifestation of patients with IBD; it can develop in up to five percent of people with Crohn’s disease or ulcerative colitis.1,4 Rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and other unspecified inflammatory arthritides are also frequently identified in patients with PG.2 These can occur even more frequently if patients also have underlying IBD.3 In addition, solid organ and hematologic malignancies and disorders, such as monoclonal gammopathy of undetermined significance, myelodysplastic syndrome, and polycythemia vera, are also associated with PG.2,7 The largest retrospective review of patients with PG (N=356) supports age-specific screening for IBD, finding that IBD was the only medical comorbidity that was more common in patients younger than 65 years of age, while all other aforementioned associated disorders were found significantly more frequently in patients 65 years of age or older.2 Importantly, though, PG might present in patients prior to presentation of the comorbidity. A recent report recommends screening for underlying disorders for a minimum of 24 months after PG diagnosis, particularly among patients younger than 50 years of age.5
As we understand more of the pathophysiology of PG, additional autoinflammatory disease associations have been recognized. PAPA syndrome is an autosomal dominant disorder characterized by the triad of pyogenic sterile arthritis, PG, and acne. PAPASH involves the tetrad of pyogenic sterile arthritis, PG, acne, and hidradenitis suppurativa, while PASH is composed of PG, acne, and hidradenitis suppurativa.1 These syndromes highlight the role of genetics and autoinflammation in the pathogenesis of PG, and investigations into these syndromes might provide further insight into more specific treatment modalities for PG.
Pathergy is an identifiable trigger in at least 30 percent of patients with PG.7 However, this should not preclude biopsy, as many other ulcerative disorders, such as infection, malignancy, and vascular disorders can mimic PG and must be ruled out.2 Characteristic PG histopathology exhibits perifollicular, neutrophilic inflammation with intradermal abscess formation.8 Binus et al7 found that only seven percent of patients with PG exhibited its classic pathology. Thus, a biopsy that is not suggestive of PG does not rule out the disorder.
PG is an immunologic, inflammatory disorder. As such, the goal of treatment is to maintain proper immunosuppression.9 There are currently no medications approved by the FDA for the treatment of PG.10 Topical therapies and gentle wound care are often insufficient to control progression of cutaneous disease; however, these therapies are an important supplement to systemic therapy. Topical and intralesional corticosteroids and calcineurin inhibitors receive the strongest support for local wound care in the literature.9 The two most commonly employed first-line systemic agents are corticosteroids and cyclosporine. The STOP GAP trial, a prospective, randomized, controlled study, compared these two most commonly used modalities and found no significant difference in healing between the treatment groups. Importantly, though, only 47 percent of patients healed, and 28 to 30 percent of patients experienced a recurrence.11
Due to the rarity of PG, few randomized, controlled trials exist. The majority of the literature on the treatment of PG are case reports or small case series. Other steroid-sparing agents, such as azathioprine, mycophenolate mofetil, and methotrexate, have been shown to have some success.9 Intralesional methotrexate has also been suggested as an effective adjuvant therapy.16 Low-dose oral tacrolimus was reported to be safe and effective in two cases of several recalcitrant perineal PG lesions.17 Antibiotics, such as minocycline, doxycycline, and dapsone, have also promoted healing, likely because of their anti-inflammatory properties.3,9 Laird et al18 reported success using apremilast in combination with prednisone and methotrexate. Other novel adjunctive treatments in the literature include platelet-rich fibrin and hyperbaric oxygen therapy.19,20 Surgical debridement and reconstructive surgery remain controversial due to the risk of pathergy, though there are reports that utilized split-thickness skin grafts and free flap reconstructions successfully.21 However, it is essential that patients remain adequately immunosuppressed during surgical procedures.
Our patient’s condition failed to respond to many of the aforementioned first- and second-line local and systemic therapies. Biologic medications were once described as third-line medications in the treatment of PG.22 However, as we better understand the major role that T-cells and cytokines play in the pathogenesis of many dermatologic diseases, including PG, we find that biologic medications are producing promising results as treatment modalities.1,4 Some researchers even suggest that TNF-alpha inhibitors should be considered a first-line therapy in patients with PG with concomitant IBD.23
Infliximab, a chimeric monoclonal TNF-alpha antibody, is currently the only biologic medication that has undergone a randomized, controlled trial for the treatment of PG.24,25 The study demonstrated a 46-percent two-week response rate in contrast with a rate of six percent in placebo patients; ultimately 69 percent of patients had a beneficial response, while 31 percent of patients had no response.25 Some studies report infliximab to be the most effective biologic for PG,23 though most investigators recognize the inconvenience of infusions and the increased risk of developing neutralizing antibodies (as high as 75%24) as disadvantages of the therapy. Infliximab might also be most prone to a loss of efficacy over time.26
Adalimumab, a fully-human monoclonal antibody against TNF-alpha, also offers promise in the treatment of PG.9 The biologic can be injected subcutaneously by the patient, in contrast with infliximab, which must be administered via infusion.27,28 Additionally, being a fully human antibody, adalimumab presents a decreased risk of neutralizing antibody development.28
Adalimumab has shown remarkable efficacy, particularly in patients with PG and concomitant IBD. The dosing and scheduling of treatment varies among cases in the literature. Table 1 lists the indications and doses of adalimumab that have shown efficacy and safety.29 The treatment of PG in the literature has followed a variety of these regimens. Some accounts report initiating a loading dose of 80mg, followed by 40mg one week later and then 40mg every other week, identical to the psoriasis and uveitis regimens. Others report a 160mg loading dose, followed by 80mg two weeks later and then 40mg every other week, which is the same as the IBD dosing schedule. There are also successful cases where treatment with 40 or 80mg was initiated weekly without a preceding loading dose.26–33 In the case of our patient, a loading dose of 160mg, followed by 80mg on Day 15, 40mg on Day 29, and weekly doses of 40mg thereafter, consistent with hidradenitis suppurativa dosing, was selected. It is difficult to evaluate the comparative efficacy of different schedules and dosing. To date, no randomized, controlled trial has been reported for the use of adalimumab in PG.

Etanercept, a fusion protein targeting TNF-alpha, has also demonstrated success in the treatment of PG; however, its efficacy in treating Crohn’s disease is minimal.9 As such, etanercept is not recommended for PG in patients with concomitant IBD. Finally, ustekinumab, a monoclonal antibody targeting IL-12 and IL-23, has also been shown to be useful in the treatment of PG.34–36 Notably, a higher dose than that used for psoriasis might be required for PG.35
With any biologic, it is essential to rule out major infections, such as tuberculosis and malignancy, prior to treatment and to monitor for adverse reactions.32 Paradoxical reactions, defined as the appearance or exacerbation of a pathological condition that ordinarily responds to this class of drug while treating a patient for another condition, have been described for PG.36,37 While rare, clinicians should be aware of the possibility of this potentially catastrophic adverse event.
Conclusion
As demonstrated by our patient, PG is notoriously refractory to treatment, particularly in cases with multiple lesions present in atypical locations, such as the face. It remains of utmost importance to rule out any other ulcerative process and to thoroughly evaluate patients for underlying systemic comorbidities. As we understand more of the pathophysiology behind PG, we can more specifically target the immunologic and inflammatory mediators contributing to the disease. Biologic medications targeting TNF-alpha, IL-23, and IL-17 offer promising new treatment modalities for PG.
References
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73(4):691–698.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154(4):409–413.
- Jiang YY, Li J, Li Y, et al. Comparison of clinical features between pyoderma gangrenosum concomitant by inflammatory bowel disease and idiopathic pyoderma gangrenosum. Chin Med J (Engl). 2017;130(22):2674–2679.
- Wang EA, Steel A, Luxardi G, et al. Classic ulcerative pyoderma gangrenosum Is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2018;8:1980.
- Vacas AS, Torre AC1, Bollea-Garlatti ML, et al. Pyoderma gangrenosum: clinical characteristics, associated diseases, and responses to treatment in a retrospective cohort study of 31 patients. Int J Dermatol. 2017;56(4):386–391.
- Powell FC, Su WP, Perry HO, et al. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34(3):395–409; quiz 410–412.
- Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165(6): 1244–1250.
- Pereira N1, Brites MM, Gonçalo M, et al. Pyoderma gangrenosum—a review of 24 cases observed over 10 years. Int J Dermatol. 2013;52(8):938–945.
- Soto Vilches F, Vera-Kellet C. Pyoderma gangrenosum: classic and emerging therapies. Med Clin (Barc). 2017;149(6):256–260.
- McKenzie F, Cash D, Gupta A, et al. Biologic and small-molecule medications in the management of pyoderma gangrenosum. J Dermatolog Treat. 2018:1–13.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Tashtoush B, Memarpour R, Johnston Y, Ramirez J. Large pyoderma gangrenosum-like ulcers: a rare presentation of granulomatosis with polyangiitis. Case Rep Rheumatol. 2014;2014:850364.
- Kurniadi I, Imanishi H, Furukawa H, et al. Case of facial pyoderma gangrenosum. J Dermatol. 2016;43(11):1373–1374.
- Kratzsch D, Ziemer M1, Milkova L, et al. Facial pyoderma gangrenosum in senescence. Case Rep Dermatol. 2013;5(3):295–300.
- Okamura K, Konno T, Onami K, et al. A case of primarily facial pyoderma gangrenosum associated with Takayasu arteritis. JAAD Case Rep. 2017;3(2):124–126.
- Del Puerto C, Navarrete-Dechent CP, Carrasco-Zuber JE, Vera-Kellet C. Intralesional methotrexate as an adjuvant treatment for pyoderma gangrenosum: a case report. Indian J Dermatol Venereol Leprol. 2017;83(2):277.
- Sadati MS, Dastgheib L, Aflaki E. Recalcitrant cases of pyoderma gangrenosum, responding dramatically to systemic tacrolimus. G Ital Dermatol Venereol. 2017;152(3):308–310.
- Laird ME, Tong LX, Lo Sicco KI, et al. Novel use of apremilast for adjunctive treatment of recalcitrant pyoderma gangrenosum. JAAD Case Rep. 2017;3(3):228–229.
- Fortunato L, Barone S, Bennardo F, Giudice A. Management of facial pyoderma gangrenosum using platelet-rich fibrin: a technical report. J Oral Maxillofac Surg. 2018;76(7):1460–1463.
- Seo HI, Lee HJ, Han KH. Hyperbaric oxygen therapy for pyoderma gangrenosum associated with ulcerative colitis. Intest Res. 2018;16(1):155–157.
- Seok HH, Kang MS, Jin US. Treatment of atypical pyoderma gangrenosum on the face. Arch Plast Surg. 2013;40(4):463–465.
- Feldman SR, Lacy FA, Huang WW. The safety of treatments used in pyoderma gangrenosum. Expert Opin Drug Saf. 2018;17(1):55–61.
- Arivarasan K, Bhardwaj V, Sud S, et al. Biologics for the treatment of pyoderma gangrenosum in ulcerative colitis. Intest Res. 2016;14(4):365–368.
- Hubbard VG, Friedmann AC, Goldsmith P. Systemic pyoderma gangrenosum responding to infliximab and adalimumab. Br J Dermatol. 2005;152(5):1059–1061.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–509.
- Fonder MA, Cummins DL, Ehst BD, et al. Adalimumab therapy for recalcitrant pyoderma gangrenosum. J Burns Wounds. 2006;5:e8.
- Sagami S, Ueno Y, Tanaka S, et al. Successful use of adalimumab for treating pyoderma gangrenosum with ulcerative colitis under corticosteroid-tapering conditions. Intern Med. 2015;54(17):2167–2172.
- Heffernan MP, Anadkat MJ, Smith DI. Adalimumab treatment for pyoderma gangrenosum. Arch Dermatol. 2007;143(3):306–308.
- AbbVie. Humira full prescribing information. Available from: https://www.rxabbvie.com/pdf/humira.pdf.
- Lipka S, Katz S, Ginzburg L. Massive pyoderma gangrenosum in a 77 year old female with Crohn’s disease responsive to adalimumab. J Crohns Colitis. 2013;7(5):427–428.
- Lee JH, Chang IK1, Lee HE, et al. Treatment of recalcitrant pyoderma gangrenosum with ulcerative colitis by adalimumab injection. Ann Dermatol. 2017;29(2):260–262.
- Pomerantz RG, Husni ME, Mody E, Qureshi AA. Adalimumab for treatment of pyoderma gangrenosum. Br J Dermatol. 2007;157(6): 1274–1275.
- Campanati A, Brisigotti V, Ganzetti G, et al. Finally, recurrent pyoderma gangrenosum treated with adalimumab: case report and review of the literature. J Eur Acad Dermatol Venereol. 2015;29(6):1245–1247.
- Greb JE, Gottlieb AB, Goldminz AM. High-dose ustekinumab for the treatment of severe, recalcitrant pyoderma gangrenosum. Dermatol Ther. 2016;29(6):482–483.
- Low ZM, Mar A. Treatment of severe recalcitrant pyoderma gangrenosum with ustekinumab. Australas J Dermatol. 2018;59(2):131–134.
- Benzaquen M, Monnier J, Beaussault Y, et al. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: effectiveness of ustekinumab. Australas J Dermatol. 2017;58(4):e270–e271.
- Puig L. Paradoxical reactions: anti-tumor necrosis factor alpha agents, ustekinumab, secukinumab, ixekizumab, and others. Curr Probl Dermatol. 2018;53:49–63.