 J Clin Aesthet Dermatol. 2023;16(4):28-31.
J Clin Aesthet Dermatol. 2023;16(4):28-31.
by Christopher J. Sayed, MD; Salma E. El-Behaedi, BS; Lindsay P. Osborn, MD;
Paul B. Googe, MD; and Franklin R. Blum, BS
Drs. Sayed and Osborn are with the Department of Dermatology at UNC Chapel Hill School of Medicine in Chapel Hill, North Carolina. Ms. El-Behaedi and Mr. Blum are also with the Department of Dermatology at UNC Chapel Hill School of Medicine in Chapel Hill, North Carolina. Dr. Googe is with the UNC Chapel Hill School of Medicine, UNC Department of Pathology and Laboratory Medicine, and the UNC Lineberger Comprehensive Cancer Center in Chapel Hill, North Carolina.
FUNDING: No funding was provided for this article
DISCLOSURES: The authors report no conflicts on interest relevant to the content of this article.
ABSTRACT: Objective. No known studies have attempted to describe the pathophysiological relationship between patients who develop both porokeratosis and hidradenitis suppurativa (HS). The purpose of this report is to present possible immunological mechanisms that predispose patients to developing both porokeratosis and HS.
Methods. In this case series, patients were identified during routine clinical encounters and data was extracted from the electronic medical record from October 2010 until April 2021. This study is a single center case series including patients from the department of dermatology at the UNC School of Medicine in Chapel Hill, North Carolina. Patients were selected via digital chart review if they had simultaneous diagnoses of disseminated porokeratosis and HS. Two eligible patients were identified as actively receiving care. One patient is a Black female and the other a White male. No primary study outcomes were planned. This investigation utilized chart review to identify disease time course, which was subsequently used to elucidate study outcomes.
Results. Patient A is a 54-year-old Black female and Patient B is a 65-year-old White male. Both patients developed porokeratosis after multiple years of living with HS. Immunosuppression with adalimumab, corticosteroids, or other medications did not clearly precede porokeratosis development in either patient.
Limitations. Limitations include that this study was conducted at a single center and prevalence of patients with concomitance of both conditions is low.
Conclusion. In patients who demonstrate simultaneous HS and porokeratosis, activation of the innate immune system and associated IL-1 production may lead to autoinflammation and a phenotype of hyperkeratinization. Mutations in genes such as mevalonate kinase may predispose subjects to the development of porokeratoses and HS.
Keywords: Hidradenitis suppurativa; porokeratosis; porokeratoses; interleukin-1 (IL-1)
Porokeratosis is a broad term used to describe multiple dermatologic disorders of hyperkeratinization with a range of clinical and phenotypic overlap,1 including porokeratosis of Mibelli, eruptive pruritic papular porokeratosis, linear porokeratosis, disseminated superficial actinic porokeratosis, and porokeratosis plantaris palmaris et disseminata (PPPD).1 PPPD is an uncommon subtype with often extensive and widespread skin involvement, including the palms and soles, which can lead to significant morbidity. The histopathologic hallmark of porokeratoses is the coronoid lamella, a compact column of parakeratotic cells with subjacent dyskeratotic keratinocytes.
Hidradenitis suppurativa (HS) is a common chronic cutaneous inflammatory disorder consisting of painful and malodorous nodules, comedones, tunnels, and scarring in intertriginous zones. Its severe impact on quality of life is well-documented,2 and associations with many comorbid conditions have been reported.3
The clinical coexistence of HS and porokeratotic conditions is largely undescribed in the literature. A few cases have described patients with disseminated porokeratoses who are incidentally reported to have a documented history of HS, but comment on the relationship between the two conditions has been limited.4,5 Each condition has been linked to hyperkeratinization, but clear linkage has not been established.6,7 This report describes two cases of patients with concurrent diagnoses of PPPD and HS and presents potential pathophysiologic links between the conditions.
Case Synopses
Case 1. Patient A is a 44-year-old Black female with a history of diabetes and eczema who presented with two years of gradually worsening extensive papules and plaques with spiny follicular prominence on the trunk (Figure 1 A-B), extremities, face, and scalp (Figure 1C). Firm hyperkeratosis was present on the palms and soles without significant scale or fissuring (Figure 1D). Previous dermatopathology reports demonstrated suppurative folliculitis and lichen simplex chronicus. Triamcinolone ointment, doxepin, adapalene gel, and oral prednisone provided minimal relief.
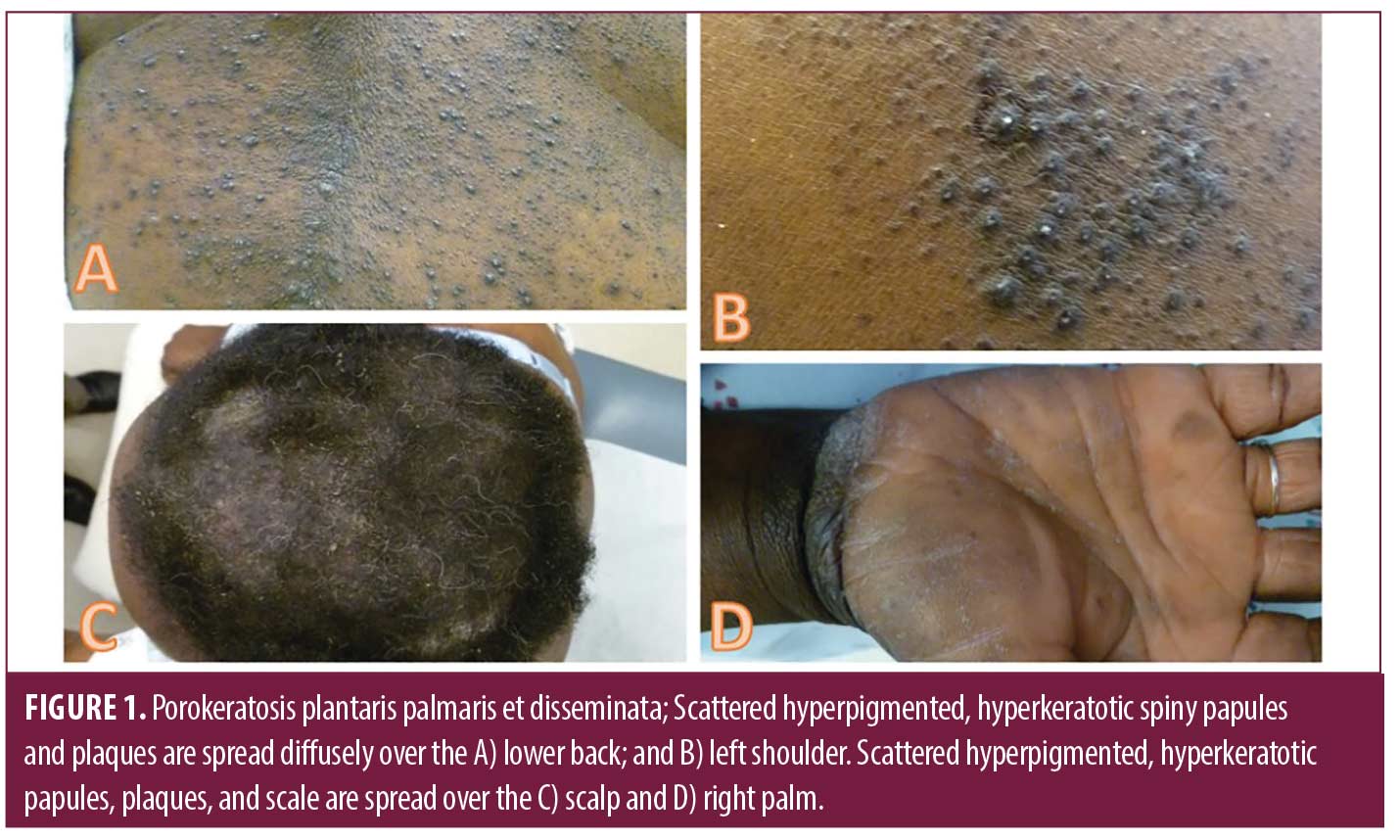
Two 4-mm punch biopsies from the right flank demonstrated several foci of coronoid lamellation, consisting of tiers of parakeratosis above dyskeratotic keratinocytes. A diagnosis of PPPD was made. Soon after, she also reported a longstanding history of recurrent abscesses and nodules of the bilateral inguinal folds and axilla that predated the findings of her PPPD, and on exam was found to have extensive scars and tunnels in the axilla, groin, and buttocks, which prompted a diagnosis of HS.
Over the subsequent 11 years, isotretinoin 40mg daily, triamcinolone 0.1% ointment, and 2% cholesterol/2% lovastatin ointment failed to improve her PPPD. Her HS has had variable and mostly inadequate responses to minocycline, doxycycline, clindamycin, moxifloxacin, rifampin, metronidazole, adalimumab 40mg subcutaneously weekly, infliximab 10mg/kg intravenously every four weeks with methotrexate 25mg orally weekly, and golimumab 200mg intravenously every four weeks. She has also had excisional procedures of the groin and buttocks for treatment of her HS. Notably, she has developed several large infundibular follicular cysts ranging in size from 4 to10 centimeters.
Case 2. Patient B is a 64-year-old White male with a history of cerebrovascular accident with residual right-sided weakness and a 20 pack-year smoking history. His initial presentation consisted of a sudden-onset pruritic eruption of the trunk, face, and extremities with upper body predominance. He then presented to our clinic two years into the course of disease with persistent, progressive involvement of the same areas. Prior dermatopathology reports suggested features of lichen amyloidosis. He had not improved with prednisone tapers, topical steroids, or oral antihistamines. He also reported a five-year history of painful nodules, abscesses, and draining tunnels of the scalp and groin that preceded the pruritic eruption by three years, which moderately improved with adalimumab 40mg weekly over the prior two years.
On physical examination, there were widespread, excoriated and lichenified plaques with follicular prominence of the trunk and extremities (Figure 2), as well as nodules, boggy plaques, and draining tunnels of the axilla, inguinal folds, and posterior occipital scalp.
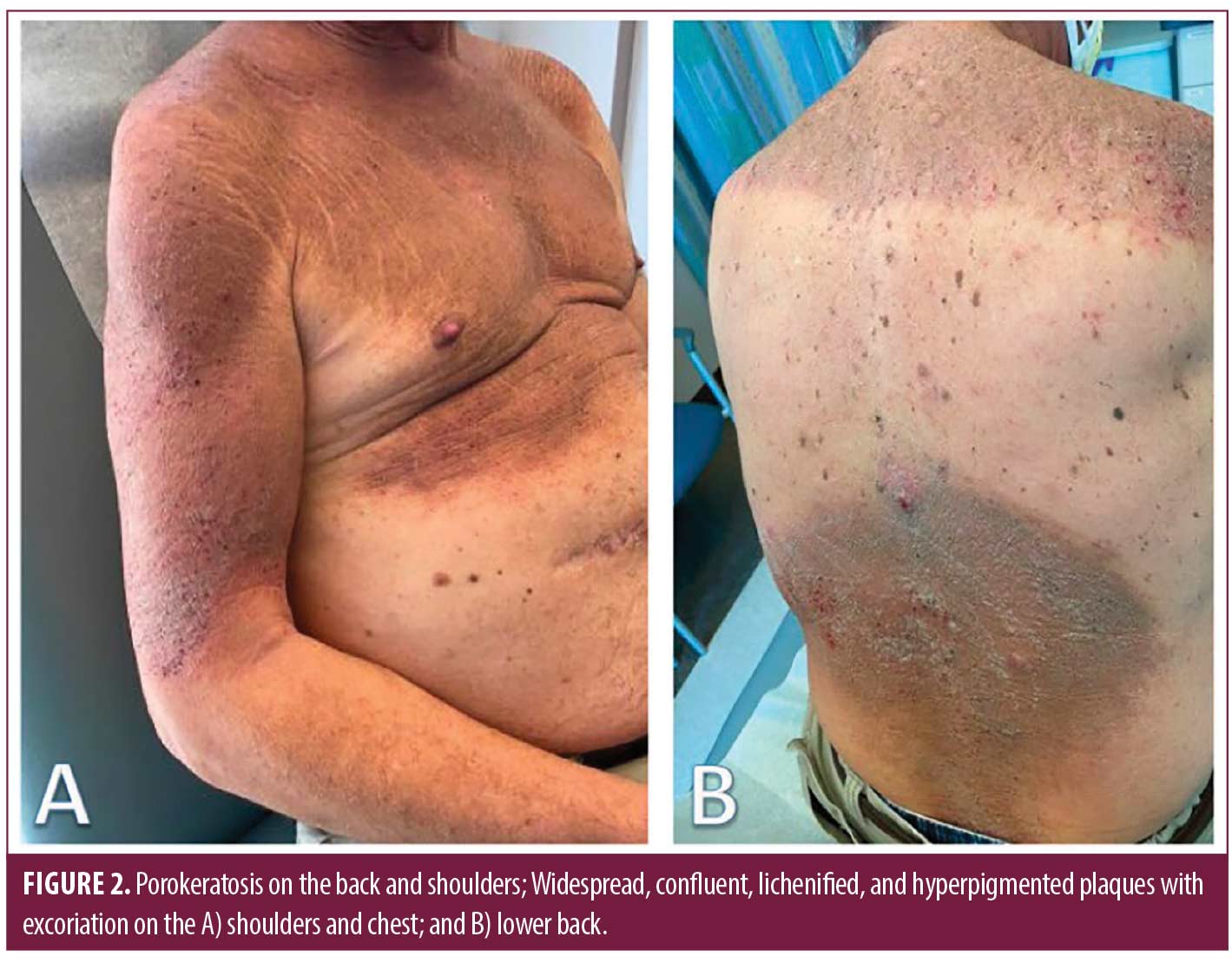
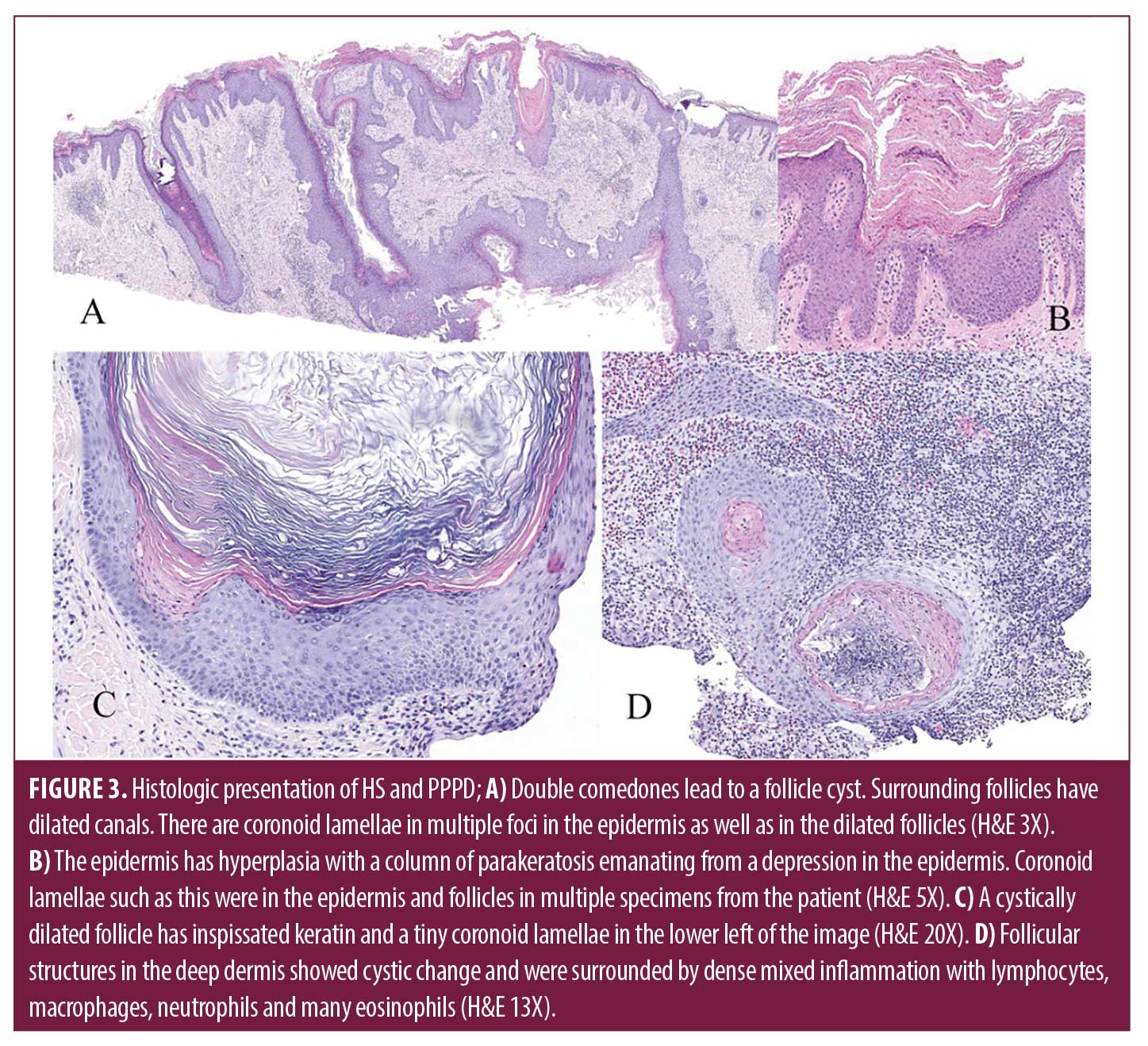
Laboratory evaluation for amyloidosis with serum protein electrophoresis was normal, and a chest x-ray was performed to rule out underlying lung cancer given his smoking history. Two 4-millimeter punch biopsies were performed on the right chest and right lower back demonstrating strikingly extensive formation of coronoid lamella and a lichenoid lympho-eosinophilic infiltrate. Five prior externally sourced biopsy specimens were also reviewed. All biopsies showed retiform epidermal hyperplasia with multifocal vertical columns of parakeratosis with subjacent dyskeratotic cells in the stratum spinosum. Many follicles had dilated canals, and some had cornoid lamellae within them. Perifollicular and perivascular infiltrates of lymphocytes and plasma cells were present. Several larger specimens had double comedones, cystically dilated follicles and foci of deep pericystic and perifollicular lymphocyte, eosinophil, and macrophage infiltrates (Figure 2). A diagnosis of disseminated porokeratosis was supported by clinicopathologic correlation. The follicular plugging, infundibular cysts, and dense inflammatory infiltrate found in HS excision specimens were consistent with HS.
Discussion
The underlying pathophysiologic link between HS and porokeratosis has yet to be fully elucidated. Akiyama et al. proposes HS and porokeratosis belong within the category of inflammatory keratinization diseases, which are characterized by autoinflammation and dysregulation of the innate immune system leading to hyperkeratosis.8 In HS, hyperkeratinization plays a central role, especially early in the disease course. Infundibular hyperkeratosis, hyperplasia of the follicular epithelium, and perifolliculitis are frequently found histologically, with one study finding hyperkeratosis of terminal follicles occurring in close to 90 percent of cases.9 Particularly in familial cases of hidradenitis where there is a clear association with other autoinflammatory diseases, upregulation of cytokine interleukin 1 (IL-1) appears to be critical to disease development.8,10 Additionally, tissue samples from patients with hidradenitis demonstrate increased expression of IL-1, caspase-1, tumor necrosis factor (TNF), and other inflammatory markers.11,12
Prior studies have identified mutations in the mevalonate kinase gene, and subsequent mevalonate kinase deficiency, as the pathophysiologic mechanism for disseminated superficial actinic porokeratosis.13,14 A more severe form of the same deficiency is also the etiology of hyperimmunoglobulin D syndrome, an autoinflammatory syndrome of the innate immune system. Interestingly, in vitro studies have demonstrated that mevalonate kinase deficiency upregulates caspase-1 activity and subsequently increases expression of IL-1,15 which is implicated in the pathogenesis of both HS and porokeratosis. Furthermore, in patients with inherent susceptibility to dysregulation of the innate immune system, increased expression of these inflammatory cytokines may result in multiple phenotypes, as seen in these two reported patients.
Examining prior reports of the two conditions, one report described a 61-year-old Caucasian woman with a past medical history of HS, Hashimoto’s thyroiditis, and a 15-year history of disseminated superficial actinic porokeratosis (DSAP) on her legs and forearms bilaterally.4 The case report discussed a novel treatment modality for DSAP but did not comment on pathophysiological links between HS and porokeratoses or her HS distribution. The second case report describes a 26-year-old man with Sturge Weber Syndrome, HS, and 8-year history of follicular porokeratosis.5 However, similarly no comment on the HS distribution or potential explanation for the presence of both HS and porokeratosis is provided in the article.
Multiple accounts describe immunosuppression leading to development of porokeratosis,16,17 including recent corticosteroid use and in transplant patients.18 Notably, development of disseminated porokeratosis following use of adalimumab has been documented.19 One explanation, therefore, is that patients with HS may be at a higher risk of developing porokeratosis due to immunosuppressive medications, which are commonly utilized in HS. Some postulate that immunosuppression might alter phenotypic expression in those with predisposing genetic mutations and trigger the expression of a mutant subset of epidermal cells by disrupting epidermal growth dynamics and promoting abnormal clone proliferation.17 For patient A, however, porokeratosis developed several years prior to the use of adalimumab or other significant immunosuppressive medications. Patient B was treated with adalimumab for two years prior to presentation at our institution. While his porokeratosis had been similarly present for about two years, he could not verify whether the rash began before or after starting adalimumab. This report does not, therefore, support medical immunosuppression as the primary pathophysiological cause of concomitant porokeratosis and HS.
In patients who demonstrate simultaneous HS and porokeratosis, activation of the innate immune system and associated IL-1 production may lead to autoinflammation and a phenotype of hyperkeratinization. Additional studies are needed to further elucidate a biomechanistic etiology of the coexistence of these diseases in some patients.
Conclusion
In patients who demonstrate simultaneous HS and porokeratosis, activation of the innate immune system and associated IL-1 production may lead to autoinflammation and a phenotype of hyperkeratinization. Mutations in genes such as mevalonate kinase may predispose subjects to the development of porokeratoses and HS.
References
- Chernosky ME. Porokeratosis. Arch Dermatol. 1986. 122(8):869–870.
- Alikhan A, et al. North American clinical management guidelines for hidradenitis suppurativa: A publication from the United States and Canadian Hidradenitis Suppurativa Foundations: Part I: Diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019. 81(1):76–90.
- Garg A, et al. Comorbidity screening in hidradenitis suppurativa: Evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2021.
- Smith J, et al Brachytherapy for Resistant Disseminated Superficial Actinic Porokeratosis. Applied Radiation Oncology. 2020. 9(3): p. 43–45.
- Young PM, et al. An unusual spiculated presentation of follicular porokeratosis. Dermatol Online J. 2019. 25(7).
- Takeichi T, et al. A novel NCSTN missense mutation in the signal peptide domain causes hidradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. Br J Dermatol. 2020. 182(2): p. 491–493.
- De Vita V and D McGonagle, Hidradenitis suppurativa as an autoinflammatory keratinization disease. J Allergy Clin Immunol. 2018.141(5): p.1953.
- Akiyama M. Autoinflammatory Keratinization Diseases (AiKDs): Expansion of Disorders to Be Included. Front Immunol. 2020.11:280.
- von Laffert M, et al. Hidradenitis suppurativa/acne inversa: bilocated epithelial hyperplasia with very different sequelae. Br J Dermatol. 2011. 164(2):367–371.
- Byrd AS, et al. Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci Transl Med. 2019. 11(508).
- Saunte, DML and Jemec GBE. Hidradenitis Suppurativa: Advances in Diagnosis and Treatment. JAMA. 2017. 318(20): 2019–2032.
- Witte-Handel E, et al. The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction. J Invest Dermatol. 2019. 139(6):1294–1305.
- Zhang SQ, et al., Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet. 2012. 44(10):1156–1160.
- Zhang Z, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015. 4:e06322.
- Normand S, et al. Specific increase in caspase-1 activity and secretion of IL-1 family cytokines: a putative link between mevalonate kinase deficiency and inflammation. Eur Cytokine Netw. 2009. 20(3):101–117.
- Raychaudhuri SP and BR Smoller. Porokeratosis in immunosuppressed and nonimmunosuppressed patients. Int J Dermatol. 1992. 31(11):781–782.
- Kanitakis J, et al., Porokeratosis and immunosuppression. Eur J Dermatol. 1998. 8(7):459–465.
- Bednarek R, et al. Eruptive disseminated porokeratosis associated with corticosteroid-induced immunosuppression. Clin Exp Dermatol. 2015.40(7):753–756.
- Frew JW and Parsi K. Adalimumab-induced porokeratosis. Australas J Dermatol. 2015. 56(3):e80-1.